Multi-Step Workflows
Jean Morrison
2023-09-21
Last updated: 2023-09-22
Checks: 2 0
Knit directory: snakemake_tutorial/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 46f2dac. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/multistep.Rmd) and HTML
(docs/multistep.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 46f2dac | Jean Morrison | 2023-09-22 | update text |
| html | c576d4c | Jean Morrison | 2023-09-22 | Build site. |
| Rmd | 30c9013 | Jean Morrison | 2023-09-22 | Add scripts: |
| html | 30c9013 | Jean Morrison | 2023-09-22 | Add scripts: |
| html | a37d414 | Jean Morrison | 2023-09-22 | Build site. |
| html | bc62309 | Jean Morrison | 2023-09-22 | updates |
| Rmd | 8972212 | Jean Morrison | 2023-09-21 | updates |
| Rmd | 656c8b4 | Jean Morrison | 2023-09-21 | add new content |
| html | 656c8b4 | Jean Morrison | 2023-09-21 | add new content |
Introduction
In this section we will expand our workflow to include multiple steps.
In this section:
- Add a rule with multiple inputs
- Add a rule with multiple outputs, use
multiext() - Execute a multi-step workflow
- Use Snakemake to draw a DAG to visualize a workflow
- Learn how Snakemake determines what to re-run
Multiple Inputs
So far our get_stats rule has one input file and one
output file. We could have either multiple inputs or multiple outputs.
These can be specified explicitly, or using utilities like
expand(). Lets add a rule that uses bcftools to concatonate
all three chromosomes together.
rule combine_data:
input: chr20 = "data/chr20.vcf.gz", chr21 = "data/chr21.vcf.gz", chr22 = "data/chr22.vcf.gz"
output: out = "data/all.vcf.gz"
shell: "bcftools concat -o {output.out} {input.chr20} {input.chr21} {input.chr22}"Notice that we have named elements in both input and
output and we can access these elements using a placeholder
by placing the name after the “.”. We can see that this specification
might get annoying so fortunately, we can still use the placeholder
{input} to access the entire list of inputs. We can also
make use of expand() instead of explicitly writing out all
of the file names. The rule above is equivalent to
rule combine_data:
input: expand("data/chr{c}.vcf.gz", c = range(20, 23))
output: "data/all.vcf.gz"
shell: "bcftools concat -o {output} {input}"If you leave rule all as we had it before, what do you
think will happen if we run Snakemake?
Multiple Outputs and multiext()
We can also have multiple outputs. Let’s write a rule that takes in
the concatonated data and uses Plink2 to perform some basic variant
quality control steps. This is a good chance to learn about another
utility that functions like expand(). Plink is going to
produce a bunch of files that all have the same root and different
extensions. To indicate this, we can use multiext().
rule variant_qc:
input: "data/all.vcf.gz"
output: multiext("data/all_varqc", ".pgen", ".psam", ".pvar", ".log")
shell: "plink2 --vcf {input} --set-missing-var-ids '@:#' --snps-only --var-min-qual 95 --geno 0.1 --maf 0.01 --make-pgen --out data/all_varqc "Finishing the workflow
Let’s add two more rules to make a workflow that computes the genetic principal components of the 100 individuals in our data. To do this, we will use Plink to write out an LD pruned list of variants. Then we will use plink to compute the PCs.
rule ld_prune:
input: multiext("data/{prefix}", ".pgen", ".psam", ".pvar")
output: "data/{prefix}.prune.in"
shell: "plink2 --pfile data/{wildcards.prefix} --indep-pairwise 1000kb 0.01 --out data/{wildcards.prefix}"
rule pca:
input: data = multiext("data/{prefix}", ".pgen", ".psam", ".pvar"), prune = "data/{prefix}.prune.in"
output: "results/{prefix}.eigenvec"
shell: "plink2 --pfile data/{wildcards.prefix} --extract {input.prune} --pca --out results/{wildcards.prefix}"Here, we have used the wildcard {prefix} to make our
rule more generic. Look at the inputs and outputs of these rules and the
previous rules. What do you expect {prefix} to be?
Finally, lets add a step that uses R to plot the results. There are
(at least) two different ways to run R with Snakemake. The first is to
use the shell: line as we have been doing. For this method,
we want an R script that we can call at the command line that takes in
the PCs and outputs a plot. It will help us keep our workflow more
generalizeable if we don’t have to hard code these file names in the R
script, so lets pass them in as arguments. Add the rule below to the
Snakefile:
rule plot_pca:
input: "results/{prefix}.eigenvec"
output: "results/{prefix}_pca.png"
shell: "Rscript code/plot_pcs.R {input} {output}"This rule makes use of a piece of code that is already in the
code/ folder which looks like this:
args <- commandArgs(trailingOnly = TRUE)
input <- args[1]
output <- args[2]
pcs <- read.table(input)
png(output, height = 4, width = 5, units = "in", res = 300)
plot(pcs$V2, pcs$V3, xlab = "PC1", ylab = "PC2", pch = 16)
dev.off()The line args <- commandArgs(trailingOnly=TRUE)
collects the command line arguments into a variable that we can access
in R.
There is an equivalent way to do this using scripts: in
place of shell:. Using the scripts: line will
cause Snakemake to directly execute a script This also works for python
and Julia scripts and can be used to knit an R Markdown file into a
report. If a piece of R code is run using scripts: we don’t
pass it arguments. Instead, it receives an S4 object named
snakemake that contains all of the parameters available in
the Snakefile. For our example, we could change the rule to read
rule plot_pca:
input: "results/{prefix}.eigenvec"
output: "results/{prefix}_pca.png"
scripts: "code/plot_pcs2.R"And write a file code/plot_pcs2.R with this content
pcs <- read.table(snakemake@input[[1]])
png(snakemake@output[[1]], height = 4, width = 5, units = "in", res = 300)
plot(pcs$V2, pcs$V3, xlab = "PC1", ylab = "PC2", pch = 16)
dev.off()Set the input in rule all to the value that you think
will execute the PCA workflow. Test your code by running a dry run and
then execute it! Remember to add the -j 1 -c 1 flags when
you execute for real.
Visualizing the DAG of jobs
To get to know our workflow better, we can use Snakemake to produce a visual representation of it. To create a png file, use
snakemake --dag | dot -Tpng > dag.pngOpen the file (or move the file to your local computer and then open it). You should see something like this
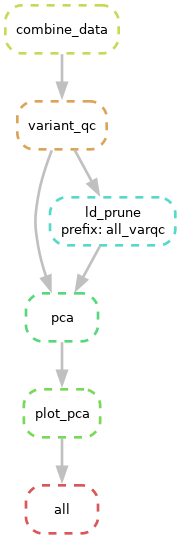
Boxes with dashed lines indicate steps that won’t be run if Snakemake were run now and solid lines indicate steps that need to be re-run.
When Rules are Re-Run
By default, Snakemake is lazy (efficient) and does not want to re-do work that has already been done. After determining the target file and the series of steps, Snakemake will check the time stamps of the intermediate products and compare these to each other and the code. If the code hasn’t been modified since files were produced, and not pre-cursors have been changed since files lower down the workflow were created, then there is nothing to do.
One way to trigger a re-run is to modify an upstream file (or to simply update it’s time stamp). To see this use
touch data/all_varqc.pgenThis will update the time stamp of data/all_varqc.pgen
to now. If you re-ran Snakemake, which steps do you think it would try
to repeat? Check your answer by running a dry-run. However, removing an
intermediate file will not trigger a re-run.
Another way to trigger a re-run is to modify the content of the rule
in the Snakefile. Note that if we are using the shell:
method to call R and we change code/plot_pcs.R, Snakemake
will not know that it needs to re-run the plotting step. However, if we
use the scripts: method to call R and change
code/plot_pcs2.R, Snakemake will detect this change and
know it should re-run.
There are several command line options that control how Snakemake
decides what to re-run. The option -F (for force) forces
Snakemake to re-run everything. Usually, this is overkill and you don’t
want to do this. The -R option can be used to tell
Snakemake to re-run everything from a given rule forward. For
example,
snakemake -j 1 -c 1 -R pcawill re-run the pca rule and the plot_pca
rule. Finally the --rerun-triggers command line option can
be used to modify the behavior of what should be re-run when. For more
details on this option, check the Snakemake documentation (this is a bit
beyond this tutorial).
Final Snakefile
At the end of this section, your Snakefile should look like this:
rule all:
input: "results/all_varqc_pca.png"
rule get_stats:
input: "data/chr{c}.vcf.gz"
output: "results/chr{c}_stats.txt"
shell: "mkdir -p results; bcftools stats {input} > {output}"
rule combine_data:
input: expand("data/chr{c}.vcf.gz", c = range(20, 23))
output: "data/all.vcf.gz"
shell: "bcftools concat -o {output} {input}"
rule variant_qc:
input: "data/all.vcf.gz"
output: multiext("data/all_varqc", ".pgen", ".psam", ".pvar", ".log")
shell: "plink2 --vcf {input} --set-missing-var-ids '@:#' --snps-only --var-min-qual 95 --geno 0.1 --maf 0.01 --make-pgen --out data/all_varqc "
rule ld_prune:
input: multiext("data/{prefix}", ".pgen", ".psam", ".pvar")
output: "data/{prefix}.prune.in"
shell: "plink2 --pfile data/{wildcards.prefix} --indep-pairwise 1000kb 0.01 --out data/{wildcards.prefix}"
rule pca:
input: data = multiext("data/{prefix}", ".pgen", ".psam", ".pvar"), prune = "data/{prefix}.prune.in"
output: "results/{prefix}.eigenvec"
shell: "plink2 --pfile data/{wildcards.prefix} --extract {input.prune} --pca --out results/{wildcards.prefix}"
rule plot_pca:
input: "results/{prefix}.eigenvec"
output: "results/{prefix}_pca.png"
shell: "Rscript code/plot_pcs.R {input} {output}"This code is in code/3.Snakefile